Fructose intolerance
With fructose intolerance, a differentiation has to be made between genetically-determined enzyme deficiency (hereditary fructose intolerance, HFI) and a fructose transport defect (fructose malabsorption). Targeted diagnostics can be carried out for both forms and clarification of the causative pathomechanism using differential diagnosis is of therapeutic relevance.
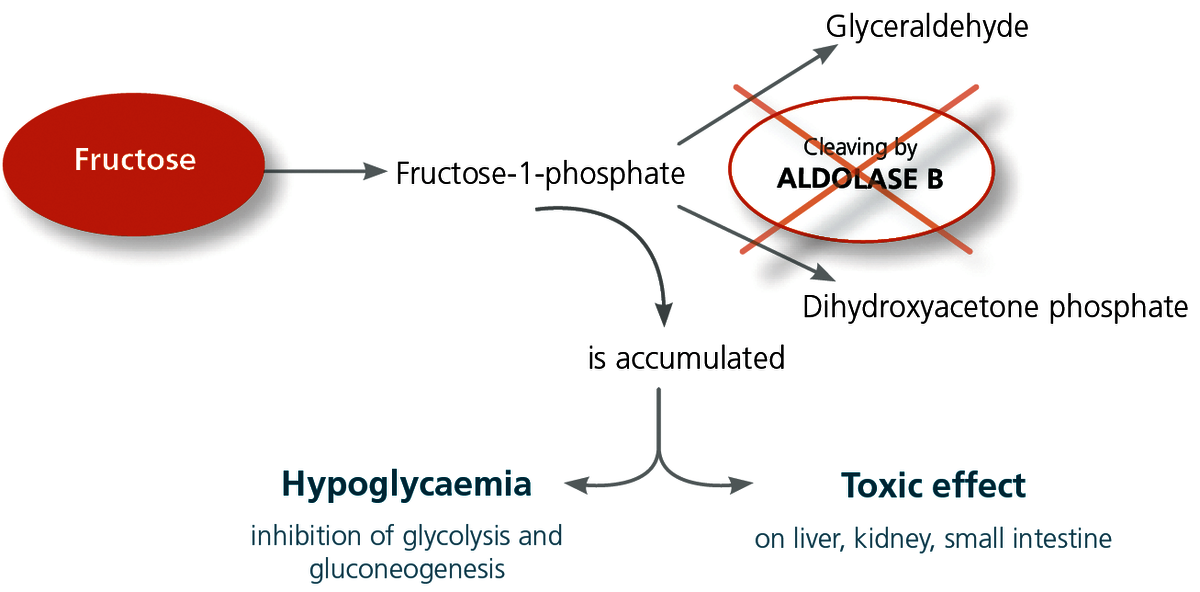
Fructose intolerance is based on an enzyme deficiency.
Fructose from the ingestion of food is metabolised in the liver. There, it is first converted to fructose-1-phosphate. The enzyme aldolase B is responsible for the next conversion stage. This enzyme is located in the cells of the liver, kidney and the mucous membrane of the small intestine, and it cleaves the fructose-1-phosphate into glyceraldehyde and dihydroxyacetone phosphate. If the aldolase B enzyme is severely impaired due to genetics, this leads to an accumulation of fructose-1-phosphate in the cells with a serious toxic effect. Furthermore, the increased fructose-1-phosphate level inhibits glycolysis.
Fructose intolerance clinical picture
HFI is a fructose metabolic disorder in which metabolites are produced that are hepatotoxic and that cause hypoglycaemia. Chronic exposure to fructose can lead to hepatomegaly and progressive hepatic insufficiency.
Babies often exhibit clinical abnormalities after they are weaned and fed with baby food that contains fructose. There may be pronounced hypoglycaemia with vomiting, attacks of sweating, neurological symptoms and even seizures, lethargy, and failure to thrive.
They often develop a dislike for sweet things and fruit, which is why it is not uncommon for a HFI to remain unnoticed into adulthood.
Symptoms of fructose intolerance (HFI)
| Acute exposure: | Chronic exposure: |
|---|---|
|
|
Fructose intolerance is genetic.
Mutations in the aldolase B gene can result in an enzyme with severely reduced activity. The pathogenic mutations responsible for an aldolase B defect can be identified with a molecular genetic test (fructose intolerance genetic test). Here, the mutations A149P, A174D, and N334K are the most common defects that occur in Europe and they are responsible for approximately 85% of all patients with HFI. The other 15% carry rarer mutations of the aldolase B gene.
The inheritance of HFI is autosomal recessive, that is, both alleles (the alleles inherited both paternally and maternally) must carry a mutation, so the enzyme deficiency is reflected clinically. The prevalence of HFI in Central Europe is approximately 1:20,000.
The diagnosis of fructose intolerance is made with a molecular genetic test.
If fructose intolerance is suspected, the test for the three most common mutations of the aldolase B gene (A149P, A174D, N334K) is carried out first. If two mutations are found, HFI is considered to be proven. If none of these more common mutations is detected, however, the probability of an HFI is low. If a single heterozygous mutation (i.e. in only one of the parental alleles) is found during the first stage, a search is carried out for other rarer mutations in the remaining areas of the aldolase B gene. A second mutation would then confirm the suspected diagnosis of an HFI.
If there is an extremely strong clinical suspicion with no verification of a mutation during the first stage, a search may also be made for other rarer mutations of the aldolase B gene in a second stage.

Therapy
The differentiation between fructose intolerance and fructose malabsorption is very important for the treatment. The only possible and successful treatment for fructose intolerance (HFI) is to avoid fructose in the diet. In the case of babies and small children up to 2-3 years of age, the diet must be strictly adhered to. With increasing age, it is possible for the tolerance of fructose to increase slightly so that the intake of fructose can be marginally adapted individually. Consistent avoidance of fructose when HFI is diagnosed early leads to a reduction of the fatty degeneration of the liver.
Essentially, with fructose intolerance, it must be remembered that many diabetic products, dietary supplements, and calorie-reduced foods contain sorbitol. As fructose is produced when sorbitol is broken down, ingesting these products can bring about life-threatening metabolic crises in affected patients.
Material required:
2 ml EDTA blood (alternatively for infants, 2 buccal swabs)
Transport of the blood sample to the laboratory is not time critical and can be done by mail. For genetic testing, we require the signed consent of the patient.For further questions please contact us by phone at 030 77001 220.
Literature
- Esposito et al. (2004): Six novel alleles identified in Italian hereditary fructose intolerance patients enlarge the mutation spectrum of the aldolase B gene. Hum Mutat. 24(6):534.
- Santer et al. (2005): The spectrum of aldolase B (ALDOB) mutations and the prevalence of hereditary fructose intolerance in Central Europe. Hum Mutat. 25(6):594.
- Wong D. (2005). Hereditary fructose intolerance. Mol Genet Metab. 85(3):165-7.